









PURPOSE
Inherited neuromuscular disorders (NMDs) are characterized by phenotypic and genotypic complexity, though the number of genes implicated in their pathology is relevant. Moreover, monogenic disorders representing orphan diseases are rare, and the co-occurrence of mutations in different causative genes is rather uncommon. The best known case of this kind is probably the co-existence of myotonic dystrophy type 2 (DM2) and myotonia congenita with CLCN1 mutation, described in several individuals [1, 2]. Other such cases presented myotonic dystrophy type 1 (DM1) and myotonia congenita with CLCN1 mutation in the same patient [3], DM1 and limb girdle syndrome [4] as well as DM1 and DM2 in one family [unpublished results]. It is postulated that the phenomenon of a co-occurrence of pathogenic variants is significantly higher among NMD phenotypes (with elevated serum creatine kinase level) compared to non-NMD phenotypes [5].
We report here on the first observation of the co- occurrence of two separate genetic entities – recessive limb-girdle muscular dystrophy type 1 (LGMDR1, previously known as LGMD2A) and DM2 in a single patient manifested by limb-girdle muscle weakness and atrophy and carrying both a homozygous CAPN3 mutation and a heterozygous CNBP expansion.
CASE DESCRIPTION
The proband, a 20-year-old male, was born to non- consanguineous parents of Polish origin. After an uneventful pregnancy, he was born by forceps delivery as a result of difficulties in passing through the birth canal due to the cross-clamping clavicle at 40 weeks of gestation, with Apgar scores of 6 and 10 points (after 1 minute) and a weight of 3700 g. His initial physical and psychomotor development was normal. As the first signs, gait and balance problems involving gross motor delay, difficulty with walking and running, tip-toe walking, and unsteadiness were noticed; he was not able to walk on his heels, jump or squat from the age of 8. The results of his electromyography (EMG) are summarized in Table 1.
Table 1
Summary of the most common manifestations of muscular dystrophy in patients with myotonic dystrophy type 2 (DM2), limb-girdle muscular dystrophy type 1 (LGMDR1), and both DM2 and LGMDR1 in the present case
His symptoms worsened significantly at the age of 14, during the period of his more rapid development; he manifested difficulties with climbing stairs, and an abnormal and waddling gait. Repeated EMG examinations of the biceps brachii muscle of the right arm primarily displayed muscle damage, whereas the sensory and motor nerve conduction velocity parameters were normal, as determined also by an EMG. At 19 years of age his general condition worsened. He experienced frequent falls and had difficulties getting up from the floor and from a chair. He complained of muscle pain during prolonged walking. On neurological assessment, a moderate proximal muscle weakness of the upper limbs, severe proximal and distal muscle weakness of the lower limbs, atrophy of the muscle of upper (proximal) and lower (thigh adductors most affected) extremities, Achilles tendon contractures, hyperlordosis, scoliosis, a positive Gowers’ sign, and foot drop were observed. No cervical abnormalities or disturbances of sensation were found. According to information from the proband’s mother, his echocardiogram was normal, though no documentation on this was available. The mother of the proband is asymptomatic; however, a slightly decreased muscle tone at age 46 years was present in neurological assessment; the father, examined at age of 48 years, was affected by diabetes mellitus from the age of 31.
The latest biochemical and morphological findings of the proband and his family members are collected in Supplementary Table 1.
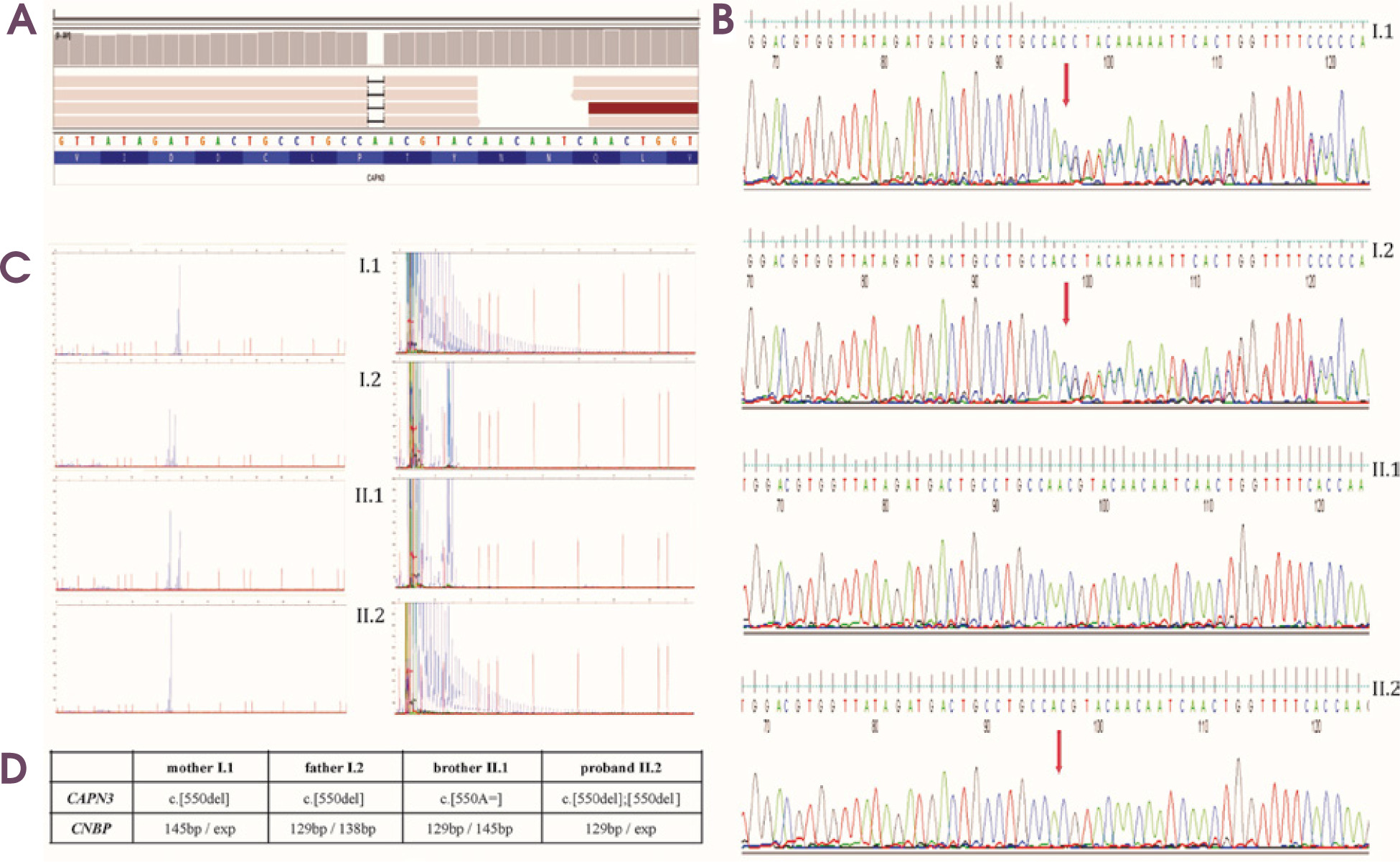
Genetic testing embraced next-generation sequencing (targeted gene panel of 89 genes, TGP) using the Illumina MiSeq instrument with MiSeq Reagent Kit v3 (150 cycles) and DM2 expansion mutation analysis. TGP sequencing enabled the identification of a homozygous frameshift pathogenic variant, NM_000070.2(CAPN3):c.550delA, p.(Thr184ArgfsTer36), in exon 4 (Figure I). After reanalysis of the sequencing the NGS data in terms of selected genes – CLCN1 and SCN4A – none of the variants in these genes were identified. Segregation analysis using Sanger sequencing identified the mother’s and father’s heterozygous carrier status, whereas the younger brother was negative for the c.550delA variant in the CAPN3. Moreover, an additional heterozygous expansion of a CCTG repeat in intron 1 of the CNBP gene, associated with DM2 in the proband, was identified. Molecular genetic analysis of family members including both parents and his brother revealed the expansion of CCTG repeats in his mother, while his father and brother displayed two normal alleles. According to the best practice guidelines and recommendations, only the qualitative result of the RP-PCR is sufficient to obtain a genetic diagnosis. Furthermore, neither the use of Southern blotting nor quadruplet- repeat primed (QP) – PCR will enable the determination of the exact sizing of the expanded repeat in DM2, due to somatic instability and cross hybridizations of the probe (in Southern blotting) and the extinction of the signal in the higher size region (in quadruplet-repeat primed [QP] – PCR).
Figure I
A) Identification a homozygous frameshift mutation in CAPN3 gene seen on Integrative Genomics Viewer software in our affected patient. B) Segregation analysis for all family members. Sanger sequencing of CAPN3 gene showing two heterozygous carriers (mother – I.1, father – I.2), one healthy brother with negative for c.550delA – II.1, and one affected homozygous patient II.2. C) Molecular analysis of family members showing expansion of CCTG repeats in the CNBP gene in mother – I.1, and affected proband – II.2, and negative myotonic dystrophy type 2 results in father – I.2, and healthy brother – II.1. D) Summary of genetic testing for all family members
DISCUSSION
As degenerative muscular conditions, LGMD primarily involve skeletal muscles, while DM involves skeletal muscle, lens, heart and liver, with an estimated worldwide incidence of 1.63 [10] and 8.26 [11] per 100,000 individuals, respectively. Therefore, it could be supposed that the co-occurrence of two rare inherited diseases may be unique. Conversely, an increased frequency of heterozygous recessive CLCN1 variants in DM2 patients with more severe and earlier myotonia (e.g., onset of grip myotonia at 14 and 20 years) has been reported, compared to exclusively DM2 patients [1, 2]. Similarly, more severe and earlier myotonia with the onset of hand cramps and difficulty relaxing the hands at age 20 years, or muscular stiffness and grip myotonia from the age of 12, have also been described; however, these were related to the presence of CNBP expansion, together with an additional mutation, or benign polymorphism in the SCN4A gene, respectively [12, 13]. It seems that both mutations and polymorphisms, in combination with a CCTG expansion, are able to modify a phenotype.
To the best of our knowledge, the case findings presented represent the first observation of the co-occurrence of LGMDR1 and DM2 in a single patient, who clinically experiences a progressive and proximal weakness similar to both entities and carries null mutations c.550delA on both alleles of the CAPN3 and a heterozygous expansion in the CNBP gene. The first one is a known founder mutation, common in the eastern Mediterranean and southeastern Europe [14]; its homozygous state is found in 29% of Polish LGMD patients [15]. It should be emphasized that the expression studies among Czech LGMD2A patients, who carried different variants including the homozygous variant c.550delA, showed a total absence of calpain-3 protein. Additionally, the type of mutation has an impact on disease onset and progression; for example, the presence of two null mutations c.550delA is associated with the early onset of symptoms while the existence of other mutations like missense and in-frame deletions are linked to later onset and milder phenotype [16].
The last follow-up assessment of the proband at 19 years revealed a moderate clinical severity of LGMD (waddling gait, Gowers’ sign, difficulty climbing stairs). In addition, it seems that foot drop is rather uncommon among LGMDR1 patients, although it has also been noted elsewhere [17]. However, an absence of DM2 symptoms (muscle myotonia, endocrine dysfunctions, cataract, and cardiac involvement) can be associated with the patient’s young age, thus these symptoms may appear later. His most recent laboratory tests revealed numerous abnormalities including slightly increased platelet distribution width, mean platelet volume, red blood cells (RBC), decreased serum high-density lipoprotein cholesterol, and serum creatinine concentrations, which are common for DM2 patients; however, most of these usually have a decreased value of RBC [18]. Elevated serum enzyme levels such as alanine aminotransferase, aspartate aminotransferase and lactate dehydrogenase were observed in both DM2 and LGMD patients [18, 19].
It is worth noting that our patient manifested typical Achilles tendon contracture leading to tip-toe walking at age 8, whereas DM2 and LGMDR1 patients typically present a proximal onset of muscle weakness of the pelvic girdle, or pelvic and scapular girdles, respectively. In addition, the presence of multiple laboratory abnormalities in our patient may be indications of an atypical phenotype.
Intriguingly, the proband’s mother with a molecularly confirmed mutation in the CNBP gene (DM2), experienced only mild hypotonia at age 46 during the most recent clinical follow-up assessment of the proband, at 19 years. She obtained numerous normal laboratory results (excluding slightly decreased eosinophils levels), although these abnormalities are more common in males [20]. Of note was the increased level of glucose and/or CK, as well as calf hypertrophy noticed in his diabetic father and brother, who are professional athletes and are negative for DM2 expansion. In addition, it seems that muscle weakness is predominantly observed in women and is associated with severe phenotype and a later onset of symptoms [20]. DM2 is characterized by a highly variable age of onset, and age or gender can modify this phenotype. In our previous experience with DM2 patients (74/207 patients) they manifested their first symptoms at over 46 years. The overall mean (± standard deviation [SD]) age of the cohort was 40.97 ± 13.76 years, with the youngest case of onset involving a 5-year-old who presented difficulties in walking and swallowing, and the oldest one a 69-year-old with muscle weakness of the extremities and EMG myotonia. The most common symptoms among the cohort mentioned were muscle weakness, myotonia ± myotonia seen on EMG, followed by cataract, muscle pain, paresis, handgrip myotonia, difficulties in walking, muscle stiffness and atrophy. It is presumed that signs of progressive weakness in the mother, as well as her son, could have appeared over time. Therefore, careful annual multi-specialized care comprising neurological, ophthalmological, cardiological, endocrinal, audiological and other evaluations of our patient should be performed. Moreover, both the proband and his mother should be monitored during the progression of the disease, and further observation is necessary to obtain final conclusions.
COMMENT
To date, several studies described the existence of genetic variants of CLCN1 and SCN4A in DM2 patients, while we report the co-occurrence of distinct and separate entities – LGMDR1 and DM2 in a single patient.
At present the patient’s phenotype corresponds with LGMDR1 rather than DM2. In his case the genetic diagnosis of both is important due to the risk of cardiovascular problems in the future.
In those populations with a relatively high prevalence of DM2, e.g. the Polish population, the co-occurrence of point mutations associated with NMD is possible and, therefore, should be taken into consideration.